热搜词:宏基因组测序 芯片检测

热搜词:宏基因组测序 芯片检测
武汉博越致和生物科技有限公司
电话:027-87705460
传真:027-87705460
地址:武汉市高新大道666号光谷生物城C6栋3楼
发布时间:2017-09-14 点击数:次
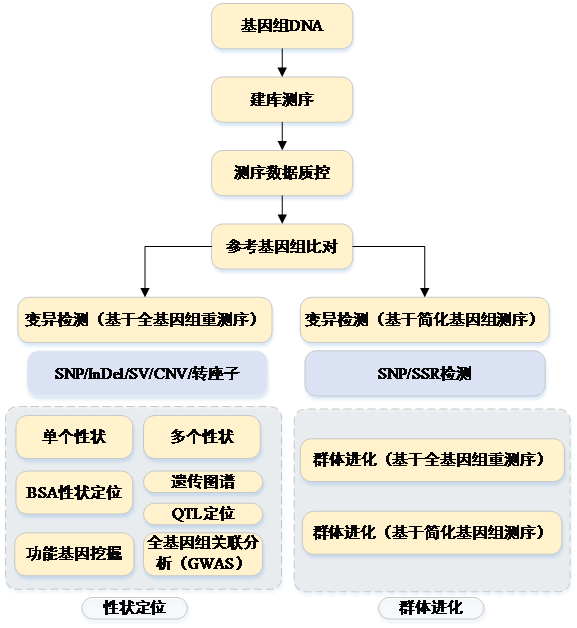
全基因组测序(Whole Genome Sequencing,WGS), 是对全基因组进行测序扫描, 能在全基因组范围内解读所有常见和稀有变异信息, 进行单核苷酸多态位点 (Single Nucleotide Polymorphisms,SNP)、插入缺失位点(Insertion Deletion,InDel)、结构变异(Structure Variation,SV)以及拷贝数变异(Copy Number Variation,CNV)深度挖掘,进而全面揭示基因组突变类型与染色体重排事件,对疾病机理研究、 药物靶点筛选等具有指导意义。 应用范围涉及临床医药研究、 群体遗传学研究、 关联分析、进化分析等众多领域。
|
样本要求 |
测序策略 |
交付周期 |
|
样品类型: 基因组DNA 样品浓度: ≥20 ng/μl(Qubit定量) 样品总量: ≥3μg(2次建库总量,不包括样品检测损耗,Qubit定量) 样品纯度: OD260/280为1.7-2.2 电泳要求: 主带清晰,无降解或轻度降解,无严重RNA、蛋白质污染(距送样日最近的电泳结果)。 |
测序模式:Illumina Hiseq Nova/MGISEQ T7 PE150 测序深度:≥30X
|
45天 |
1. Huang X, Kurata N, Wei X, et al. A map of rice genome variation reveals the origin of cultivated rice[J]. Nature, 2012, 490(7421):497-501.
