热搜词:宏基因组测序 芯片检测

热搜词:宏基因组测序 芯片检测
武汉博越致和生物科技有限公司
电话:027-87705460
传真:027-87705460
地址:武汉市高新大道666号光谷生物城C6栋3楼
发布时间:2018-10-16 点击数:次
第三代单分子测序技术经过近些年的不断开发与技术革新逐步迈向成熟。在大型基因组领域,随着单分子测序读长的不断提升、测序准确性的持续升级以及数据处理算法的优化。三代测序技术将大型基因组的组装指标Contig N50从kb级提升到Mb级,对于复杂区域、GC异常区域、结构变异的精准分析提供了坚实的保障。

超长读长,平均读长可达20kb,最长读长可达70kb,Subread Length N50可达20kb;
无GC偏好,全程无需PCR扩增;
可实时检测基因组上的碱基修饰情况,如6-mA,4-mC等;
实验背景:
“华夏一号”(Hx1)是利用PacBio测序平台绘制完成的首例亚洲人“金标准”参考基因组。众所周知,随着二代测序成本的降低,个人基因组测序已经非常普遍,但由于测序技术条件及人类基因组序列高度复杂等因素的限制,已有人类参考基因组中大量的结构变异、重复序列及GC含量异常等区域很难被准确测序,从而使得这些基因组序列缺失了大量信息,限制了后续的相关研究。借助PacBio超长读长的优势,克服了海量数据产出、大型基因组纯三代组装分析等一系列技术难点,顺利解决了上述基因组测序难题,并最终将亚洲人参考基因组组装水平提升了2个数量级,为中国人群基因组学研究、遗传疾病研究、精准医疗应用等研究领域均提供了重要的科学及临床参考信息。
实验设计:
该项目提取的样本来自一个具有正常核型的中国健康成年人新鲜的血液,利用PacBio RSII 完成基因组及全长转录组测序,选用最佳的P6-C4试剂组合,最终获得高质量、超长序列的DNA原始测序数据。此外,“华夏一号”基因组组装还结合了BioNano光学图谱分析技术,通过两种数据的整合,最终得到近完成图级别的人类基因组图谱。
实验结果:
(1)基于PacBio测序产生的数据覆盖度超过103x,BioNano产生的数据覆盖度超过101x。通过de novo组装最终得到“华夏一号”的基因组大小为2.9Gb,含有5843个Contigs,Contig N50为8.3Mb,Scaffold N50为22Mb(表1)。与已发布的其他个人基因组相比,“华夏一号”的Contig N50提高了将近10倍,且基于良好的基因组组装结果,分别比较、检测了“华夏一号”与亚洲人和白种人基因组之间的单核苷酸变异(SNVs),发现“华夏一号”与两个亚洲人基因组共有更多的SNVs,而与白种人基因组差异则较大,这与预期的研究结果是非常一致的(图1)。
(2)获得基因组完成图后,我们对Illumina和PacBio测序数据的覆盖度进行了比较,发现在GC含量异常区域,Illumina数据的覆盖度存在急剧降低的问题,而PacBio数据受到的影响则非常小(图2)。在许多Illumina数据覆盖不到(覆盖率小于5)的地方,PacBio数据都能覆盖到,这样保证了基因组组装在复杂区域的准确性和全面性(图2)。由于PacBio测序技术无需PCR扩增,基本没有GC偏向性,且其错误率又是随机产生的,因此在基因组组装上有非常明显的优势。
(3)基于PacBio测序技术获得了“华夏一号”的全长转录本,我们通过构建4个不同的文库(1-2kb、2-3kb、3-5kb和>5kb),测了共50个SMRT cells。通过对比分析由Illumina的RNA-Seq数据和PacBio全长转录组的测序数据发现,很多在Illumina测序数据中没有发现的转录本在PacBio测序数据中均可以找到,且这些转录本均能被Sanger测序方法验证。
表1. “华夏一号”与其他已公布的人类基因组序列比较
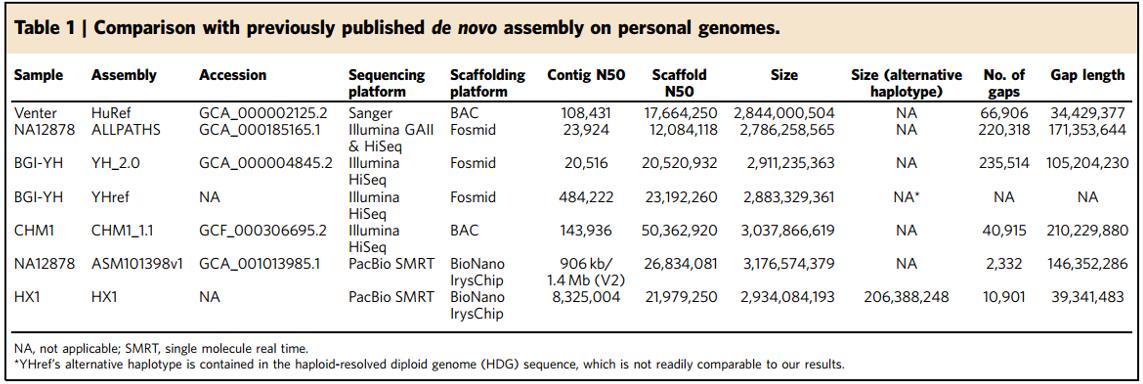
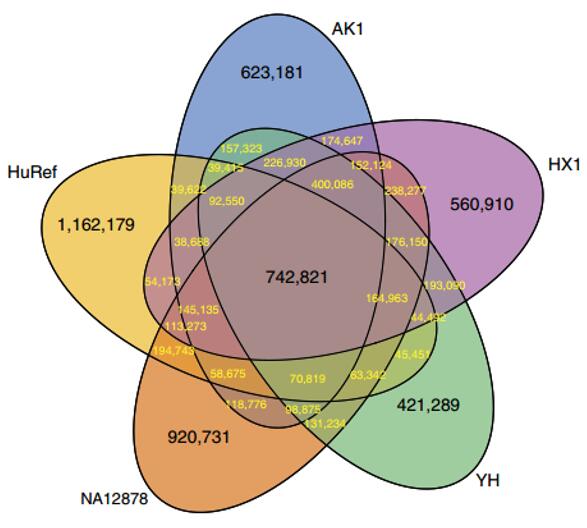
图1.不同基因组(HX1、AK1、HuRef、NA12878、YH)间共有及特有的SNVs
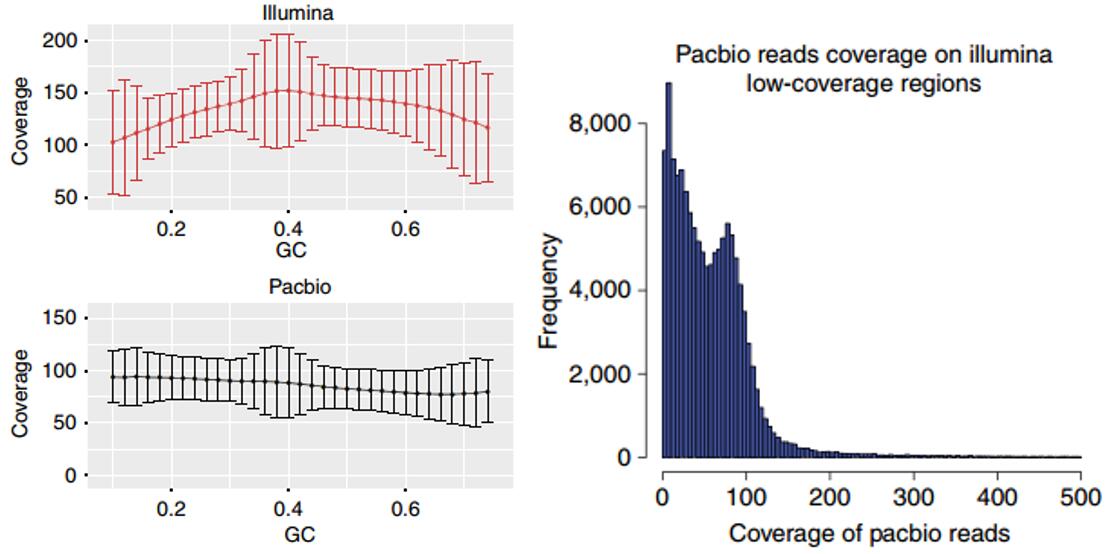
图2. Illumina和PacBio数据在GC含量异常区域中测序覆盖度(左:Illumina和PacBio数据在GC含量异常区域中测序覆盖度比较;右:PacBio覆盖到Illumina数据覆盖率小于5的区域分布)
Shi L, Guo Y, Dong C, et al. Long-read sequencing and de novo assembly of a Chinese genome[J]. Nature Communications, 2016, 7:12065.
